 首页
首页 400-620-6333
400-620-6333
DNA Damage and Repair

DNA DAMAGE AND REPAIR MECHANISMS
The formation of mutations and the development of cancer both involve damage to a cell's DNA. DNA in human cells is damaged thousands to millions of times a day by both external (exogenous) and internal metabolism (endogenous). Changes in a cell's genome can lead to errors in the transcription and subsequent translation of DNA into proteins essential for signaling and cell function. If a genome mutation is not repaired before mitosis, the mutation may also be carried into daughter cells. Once the cell is unable to effectively repair damaged DNA, three possible responses occur (see Figure 1).
?
1.?Cells may senescence, or enter an irreversible state of dormancy. In 2005, several laboratory studies found that cancer cells can senescence, stop mitosis and prevent further cell evolution in both in vivo and in vitro experiments.?1-41.
2.?Cells may die. Sufficient DNA damage initiates an apoptotic signaling cascade that forces cells into cell death.
3.?Cells become malevolent, which means they take on immortal properties and start dividing uncontrollably.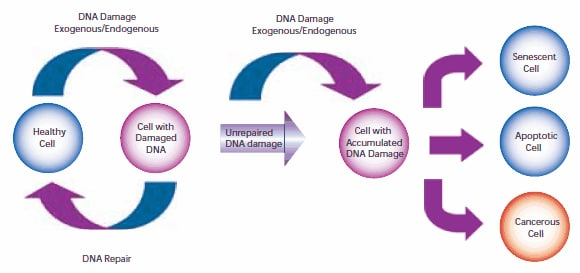
In order to compensate for the different degrees and types of DNA damage that have occurred, cells will carry out a variety of different repair procedures, including mismatch, base excision and nucleotide excision repair mechanisms, among which there is basically no redundancy processing. If there is too much damage to repair effectively by expending energy, the cell can enter the process of senescence or apoptosis. The odds that a cell will repair damage effectively depend on the type of cell and its age.
SOURCE ?OF DNA DAMAGE
For many years, exogenous damage has been considered as the main factor causing carcinogenic DNA mutations. However, Jackson and Loeb argue that endogenous DNA damage also contributes significantly to carcinogenic mutations.5 ?Environmental and cellular triggers can cause similar DNA damage.
?
DNA can be attacked by both physical and chemical mutagens. Physical mutagens are primarily a variety of radiation sources, including UV (200-300 nm wavelength) radiation from the sun. UV radiation can form covalent bonds between the bases of adjacent pyrimidines (cytosine and thymine) in the DNA strand, resulting in cross-linking. Ionizing radiation (X-rays) generates free radicals in cells, which in turn produces reactive oxygen species (ROX), causing single and double strand breaks in the double helix structure and triggering DNA mutations. Chemical mutagens can covalently attach alkyl groups to DNA bases; Nitrogen mustard compounds can be methylated or ethylated DNA bases, is a DNA alkylation agent. Precarcinogens are chemically inert precursor substances that can be transformed into highly active carcinogens through metabolism. These carcinogens can react with DNA to form DNA adducts, chemical entities that attach to DNA. Benzo [a] pyrene, a polycyclic aromatic hydrocarbon that is not carcinogenic in itself, is produced by two successive oxidation reactions under the enzyme cytochrome P450 to form epoxide Benzo [a] pyrene (BPDE), a carcinogenic metabolite that forms covalent DNA adduces (see Figure 2).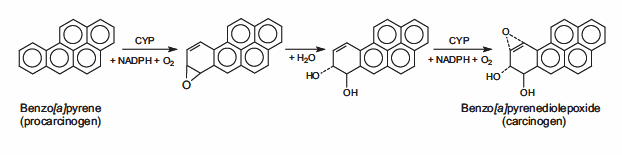
DNA damage may also result from endogenous metabolism or biochemical reactions, some of which are not well understood. Hydrolysis reactions can cut nucleotide bases from DNA strands in part or in whole. The chemical bonds that connect the purine bases (adenine or guanine) to the deoxyribonucleic acid chains spontaneously break in a process called depurination. In mammalian cells, an estimated 10,000 depurine events occur every day.?7?Depyrimidine (loss of pyrimidine bases from thymine or cytosine) is 20 to 100 times less likely to occur than depurine.
?
Deamination refers to the loss of amino groups in the adenine, guanine, and cytosine rings within the cell to hypoxanthine, xanthine, and uracil, respectively. DNA repair enzymes recognize and correct these abnormal bases. However, uncorrected uracil may be misread as thymine during subsequent DNA replication, resulting in a C→T point mutation.
?
DNA methylation is a special form of alkylation caused by S-adenosine methionine (SAM) reactions in cells. SAM is an intracellular metabolic intermediate containing highly active methyl groups. In mammalian cells, methylation occurs at position 5 of the cytosine ring of the cytidine base (C), corresponding to 5 'of the guanine base (G), the sequence CpG. Spontaneous deamination of the methylated product 5-methylcytosine is a major source of mutational errors. The loss of the amino group produces a thymine base, which is not recognized as abnormal by DNA repair enzymes, allowing the substitution to be preserved in DNA replication, resulting in a C→T point mutation (see Figure 3).
?
Figure 3.?Second-stage mutation of cytosine gives rise to thymine, which forms a C→T point mutation.
Normal metabolic processes produce reactive oxygen species (ROS), which modify bases through peroxide reactions. The purine and pyrimidine bases are oxidized. The most common mutation is the oxidation of guanine to 8-oxy-7, 8-dihydroguanine, resulting in the formation of the nucleotide 8-oxy-deoxyguanosine (8-oxo-dG). 8-oxo-dG can pair with the deoxyadenosine base instead of the expected deoxycytidine. If this error is not detected and corrected by the mismatch repair enzyme, then the DNA copied later will contain A C→A point mutation. ROS may also cause DNA depurine, depyrimidine, and single or double strand breakage.
?
DNA replication during the S phase of the cell cycle may introduce other types of genetic mutations. The polymerase that copies the DNA template has a small but significant error rate, comparing the template DNA to integrate the wrong nucleotides into the synthetic strand according to Watson-Crick pairing. Chemically altered nucleotide precursors may also be incorporated into synthetic DNA by polymerases, replacing normal bases. In addition, polymerase is prone to "slippage" when replicating DNA fragments containing large amounts of repeating amino acids or repeating sequences (microsatellite regions). This enzymatic "slippage" occurs when the template DNA strand and the copied DNA strand slip out of the correct alignment. The result is that the polymerase cannot insert the specified number of nucleotides in the template DNA correctly, resulting in too many or too few nucleotides in the daughter chain.
?
Single and double strands of DNA can break. The break of the single strand may be caused by damage to the deoxyribose part of the DNA deoxyribose phosphate chain. A break may also occur in the intermediate step of the base excisement repair path after removal of the deoxyribonucleic phosphate group by AP-endonuclease?1. 8?When a single strand break occurs, both the nucleotide base and the deoxyribose skeleton are lost from the DNA structure. Double-strand breaks occur most often during the S phase of cell passage, when DNA uncoils to serve as a template for replication, making it more likely to break.
?
DNA REPAIR MECHANISM
Cells can evolve to a state of apoptosis or senescence, which can be regarded as the ultimate means of the cell. For different types of DNA damage, cells evolve specific ways to repair the damage or remove damaging compounds.
?
O6-methylguanine DNA methyltransferase (MGMT; DNA alkyl transferase) excises methyl and ethyl adducts from guanine bases of DNA structure. This reaction is not a catalytic (enzymatic) reaction, but a stoichiometric (chemical) reaction in which one molecule MGMT is consumed for each adducts removed. The modified MGMT overexpressed cells are more tolerant to cancer, possibly because they can counteract more alkyl damage. A recent study by Niture et al. showed that MGMT expression could be improved using cysteine/glutathione enhanced drugs and natural antioxidants.?9
?
DNA polymerases with corrective activity, such as polymerase-δ, are mainly involved in replication mismatch repair. When a mismatch is detected, these polymerases stop the DNA replication process, go back to the eliminator nucleotides until the mismatch nucleotides are eliminated, and then resume the positive replication process. The data showed that mice with point mutations in both copies of the Pold1 gene showed a loss of DNA polymerase-δ correction activity and a significantly increased incidence of epithelial tumors compared with those with wild-type or single-copy mutations.?10
?
A group of proteins called mismatch excision repair (MMR) enzymes can correct errors in the corrected activity of DNA polymerase during replication. The MMR enzyme excizes faulty nucleotides in the daughter strand DNA and repairs the strand by W-C pairing, using the parent strand DNA as the correct template.?11?This is particularly important for errors arising from replication in microsatellite regions, which cannot be detected by the proofreading activity of DNA polymerase. To a limited extent, MMR enzymes can correct a variety of base pairing abnormalities caused by DNA oxidation or alkylation. These mutations include modified base pairs containing O6-methylguanine and 8-oxyguanine, carcinogens and cisplatin adducts.?12,13?Mutations in human mismatch excision repair genes MSH2 and MLH1 are associated with inherited non-polyposis colorectal cancer (HNPCC) syndrome.?14
BASE EXCISION REPAIR AND NUCLEOTIDE EXCISION REPAIR
Base excision repair (BER) involves a variety of enzymes to remove and replace a single damaged nucleotide base. BER enzymes repair base modifications caused by endogenous oxidation and hydrolysis. DNA glycosylase cleaves the chemical bond between nucleotide bases and ribose, resulting in a complete DNA ribophosphoric acid chain, but produces purine-free or pyrimidine-free (AP) sites. 8-oxyguanine DNA glycosylase (Ogg1) removes 7, 8-dihydro-8-oxyguanine (8-oxoG), a base mutation caused by reactive oxygen species. Polymorphisms in the human OGG1 gene are associated with the risk of several cancers, such as lung and prostate cancer. Uracil DNA glycosylase is another BER enzyme that excies the uracil produced by cytosine deamination, thereby preventing subsequent C→T point mutations. 15N-methylpurine DNA glycosylase (MPG) can remove a large number of modified purine bases.?16
Ap-endonucrenase 1 (APE1) can repair the AP sites in DNA produced by BER enzyme reactions and by depyrimidine and depurine processes. APE1 can cut the 5 'position of the AP site phosphodiester chain, causing the DNA chain to produce 3' -hydroxyl group and 5 '-base deoxyribonucleophosphate group. DNA polymerase β (Polβ) inserts the correct nucleotide based on the corresponding W-C pairing and removes the deoxyribose phosphate group through associated AP lyase activity. The presence of X-ray repair cross complementary Group 1 (XRCC1) is necessary for DNA ligase III (LIG3) to form heterodimers. XRCC1 acts as a scaffold protein, providing an inactive binding site for Polβ and bringing Polβ to the repair site along with LIG3 enzymes.?17?Poly (ADP-ribose) polymerase (PARP-1) interacts with XRCC1 and Polβ and is an essential component of the BER pathway.?18,19?The final step of repair is done by LIG3, which connects deoxyribose, which is used to replace nucleotides, to the deoxyribose phosphate skeleton. This fix is called "short patch BER".?20
?
Another repair path, called Long Patch BER, can replace nucleotide chains with a minimum length of two nucleotides. The pathway has been reported to repair nucleotide chains 10 to 12 nucleotides in length.?21,22?long patch BER requires proliferating nuclear antigen (PCNA) as a scaffold protein for reconstructing enzymes.?23?Other DNA polymerases, possibly polδ and Polε,?24?are used to generate oligonucleotide flaps. The existing nucleotide sequence is removed by FEN1, and oligonucleotides are then attached to the DNA via DNA ligase 1 (LIG1) to fill in the gap and complete repair.?17?The mechanism of short patch BER and long patch BER path selection is still under study (see Figure 4).?25
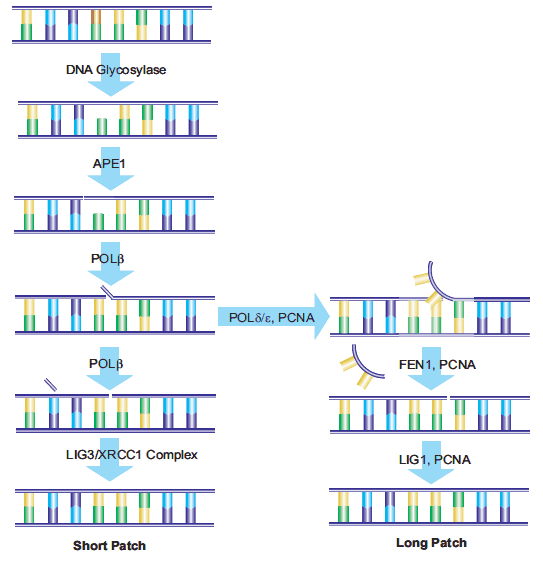
Figure 4.?Schematic diagram of short patch and long patch paths.
Although BER can displace multiple nucleotides via the long patch pathway, both short patch and long patch BER are caused by damage to a single nucleotide, thereby minimizing the impact on the DNA double helix structure. Nucleotide excisational repair (NER) repairs damage to nucleotide chains (containing at least 2 bases) that have distorted the DNA structure. In addition to repairing continuity damage caused by larger DNA adducts and exogenous factors of UV rays, NER can also repair single-strand breaks. 26?This pathway can also be used to repair damage caused by oxidative stress. 27?More than 20 proteins are involved in the NER pathway in mammalian cells, including XPA, XPC-hHR23B, replicating protein A (RPA), transcription factors TFIIH, XPB and XPD DNA helicases, ERCC1-XPF and XPG, Polδ, Polε, PCNA, and replicator C. Overexpression of the 28?excision repair Cross complementarity (ERCC1) gene is associated with cisplatin tolerance in non-small cell lung cancer cells, and 29?enhances DNA repair. 30?Genome-wide NER (GGR) can repair damage throughout the genome, and a specific NER pathway called transcription coupled repair (TCR) can repair genes during active RNA polymerase transcription. 31
DOUBLE STRAND BREAK REPAIR
Double-strand breaks in DNA can lead to loss and rearrangement of gene sequences. These fractures can be repaired by either nonhomologous end-junction (NHEJ) or homologous recombination (HR), also known as recombination repair or template-assisted repair.
?
The HR pathway is activated when the cell is late in the S/G2 phase and the template has recently been replicated. This mechanism requires an identical or nearly identical sequence connected to the damaged region of DNA via the centromere as a repair template. Double-strand breaks repaired by this mechanism are usually caused by the replicator attempting to synthesize over the broken site of the single strand or unrepaired damage, resulting in the collapse of the replicator fork structure.
At other points in the cell cycle, the non-homologous terminal junction (NHEJ) pathway is activated when sister chromosomes fail to serve as HR templates. When a break occurs, the cell does not copy the region of DNA that contains the site of the break, so unlike the HR pathway, no corresponding template strand is available. In the NHEJ pathway, the Ku heterodimer protein is located at either end of the broken DNA strand and is repaired without a template available, so sequence information may be lost in the process. Several enzymes are involved in the reconnection process, including DNA ligase IV, XRCC4, and DNA-dependent protein kinase (DNA-PK). 32,33NHEJ is inherently mutagenic because it relies on the opportunistic pairing of the ends of the single strand of two pieces of DNA that need to be joined, called microhomology (see Figure 5). In advanced eukaryotic cells, DNA-PK is required for NHEJ repair, both as the primary mechanism and as an alternative standby mechanism (D-NHEJ). 34

Figure 5.?The conventional mechanism of NHEJ repairing DNA double-strand breaks.
FURTHER APPLICATION
Although DNA damage is a key factor in the development and evolution of cancer cells, continuous damage is still used as part of clinical cancer treatment to force malignant cells into a state of apoptosis or senescence. Many chemotherapeutic drugs, such as bleomycin, mitomycin and cisplatin, are effective because they cause further DNA damage to cancer cells that replicate faster than surrounding tissue. The cellular DNA repair mechanism is a double-edged sword. On the one hand, it can reduce carcinogenic mutations and thus ensure genomic integrity, but on the other hand, in malignant cells, the same mechanism prevents the cell from further DNA damage and continues to grow uncontrollably. To block this survival mechanism within cancer cells, clinical trials are now using inhibitors of specific DNA repair enzymes, including MGMT, PARP, and DNA-PK. 35-38
References
1.Collado?M,?Gil?J,?Efeyan?A,?Guerra?C,?Schuhmacher?AJ,?Barradas?M,?Benguría?A,?Zaballos?A,?Flores?JM,?Barbacid?M,?et al.?2005.?Senescence in premalignant?tumours.?Nature.?436(7051):642-642.?https://doi.org/10.1038/436642a
2.Chen?Z,?Trotman?LC,?Shaffer?D,?Lin?H,?Dotan?ZA,?Niki?M,?Koutcher?JA,?Scher?HI,?Ludwig?T,?Gerald?W,?et al.?2005.?Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient?tumorigenesis.?Nature.?436(7051):725-730.?https://doi.org/10.1038/nature03918
3.Michaloglou?C,?Vredeveld?LCW,?Soengas?MS,?Denoyelle?C,?Kuilman?T,?van der Horst?CMAM,?Majoor?DM,?Shay?JW,?Mooi?WJ,?Peeper?DS.?2005.?BRAFE600-associated senescence-like cell cycle arrest of human naevi.?Nature.?436(7051):720-724.?https://doi.org/10.1038/nature03890
4.Braig?M,?Lee?S,?Loddenkemper?C,?Rudolph?C,?Peters?AH,?Schlegelberger?B,?Stein?H,?D?rken?B,?Jenuwein?T,?Schmitt?CA.?2005.?Oncogene-induced senescence as an initial barrier in lymphoma development.?Nature.?436(7051):660-665.?https://doi.org/10.1038/nature03841
5.Jackson?AL,?Loeb?LA.?2001.?The contribution of endogenous sources of DNA damage to the multiple mutations in cancer.?Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis.?477(1-2):7-21.?https://doi.org/10.1016/s0027-5107(01)00091-4
6.De Bont?R.?2004.?Endogenous DNA damage in humans: a review of quantitative data.?Mutagenesis.?19(3):169-185.?https://doi.org/10.1093/mutage/geh025
7.Lindahl?T,?Nyberg?B.?1972.?Rate of depurination of native deoxyribonucleic acid.?Biochemistry.?11(19):3610-3618.?https://doi.org/10.1021/bi00769a018
8.Brem?R.?2005.?XRCC1 is required for DNA single-strand break repair in human cells.?Nucleic Acids Research.?33(8):2512-2520.?https://doi.org/10.1093/nar/gki543
9.Niture?SK,?Velu?CS,?Smith?QR,?Bhat?G,?Srivenugopal?KS.?Increased expression of the MGMT repair protein mediated by cysteine prodrugs and chemopreventative natural products in human lymphocytes and tumor cell lines.?Carcinogenesis.?28(2):378-389.?https://doi.org/10.1093/carcin/bgl155
10.Goldsby?RE,?Hays?LE,?Chen?X,?Olmsted?EA,?Slayton?WB,?Spangrude?GJ,?Preston?BD.?2002.?Nonlinear partial differential equations and applications: High incidence of epithelial cancers in mice deficient for DNA polymerase ? proofreading.?Proceedings of the National Academy of Sciences.?99(24):15560-15565.?https://doi.org/10.1073/pnas.232340999
11.Yang?W.?2000.?Structure and function of mismatch repair proteins.?Mutation Research/DNA Repair.?460(3-4):245-256.?https://doi.org/10.1016/s0921-8777(00)00030-6
12.Iyer?RR,?Pluciennik?A,?Burdett?V,?Modrich?PL.?2006.?DNA Mismatch Repair:? Functions and Mechanisms.?Chem. Rev..?106(2):302-323.?https://doi.org/10.1021/cr0404794
13.Modrich?P.?2006.?Mechanisms in Eukaryotic Mismatch Repair.?J. Biol. Chem..?281(41):30305-30309.?https://doi.org/10.1074/jbc.r600022200
14.Müller?A,?Fishel?R.?2002.?Mismatch Repair and the Hereditary Non-polyposis Colorectal Cancer Syndrome (HNPCC).?Cancer Investigation.?20(1):102-109.?https://doi.org/10.1081/cnv-120000371
15.Lindahl?T.?1974.?An N-Glycosidase from Escherichia coli That Releases Free Uracil from DNA Containing Deaminated Cytosine Residues.?Proceedings of the National Academy of Sciences.?71(9):3649-3653.?https://doi.org/10.1073/pnas.71.9.3649
16.Singer?B,?Hang?B.?1997.?What Structural Features Determine Repair Enzyme Specificity and Mechanism in Chemically Modified DNA??.?Chem. Res. Toxicol..?10(7):713-732.?https://doi.org/10.1021/tx970011e
17.Lindahl?T.?1999.?Quality Control by DNA Repair.?286(5446):1897-1905.?https://doi.org/10.1126/science.286.5446.1897
18.Caldecott?KW,?Aoufouchi?S,?Johnson?P,?Shall?S.?1996.?XRCC1 Polypeptide Interacts with DNA Polymerase ? and Possibly Poly (ADP-Ribose) Polymerase, and DNA Ligase III Is a Novel Molecular 'Nick-Sensor' In Vitro.?Nucleic Acids Research.?24(22):4387-4394.?https://doi.org/10.1093/nar/24.22.4387
19.Dantzer?F,?de la Rubia?G,?Ménissier-de Murcia?J,?Hostomsky?Z,?de Murcia?G,?Schreiber?V.?2000.?Base Excision Repair Is Impaired in Mammalian Cells Lacking Poly(ADP-ribose) Polymerase-1?.?Biochemistry.?39(25):7559-7569.?https://doi.org/10.1021/bi0003442
20.Srivastava?DK,?Vande Berg?BJ,?Prasad?R,?Molina?JT,?Beard?WA,?Tomkinson?AE,?Wilson?SH.?1998.?Mammalian Abasic Site Base Excision Repair.?J. Biol. Chem..?273(33):21203-21209.?https://doi.org/10.1074/jbc.273.33.21203
21.Ranalli?TA,?Tom?S,?Bambara?RA.?2002.?AP Endonuclease 1 Coordinates Flap Endonuclease 1 and DNA Ligase I Activity in Long Patch Base Excision Repair.?J. Biol. Chem..?277(44):41715-41724.?https://doi.org/10.1074/jbc.m207207200
22.Sattler?U,?Frit?P,?Salles?B,?Calsou?P.?2003.?Long?patch DNA repair synthesis during base excision repair in mammalian cells.?EMBO Rep.?4(4):363-367.?https://doi.org/10.1038/sj.embor.embor796
23.Fortini?P,?Pascucci?B,?Parlanti?E,?Sobol?RW,?Wilson?SH,?Dogliotti?E.?1998.?Different DNA Polymerases Are Involved in the Short- and Long-Patch Base Excision Repair in Mammalian Cells?.?Biochemistry.?37(11):3575-3580.?https://doi.org/10.1021/bi972999h
24.Klungland?A.?1997.?Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1).?16(11):3341-3348.?https://doi.org/10.1093/emboj/16.11.3341
25.Sung?J,?Demple?B.?2006.?Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA.?FEBS Journal.?273(8):1620-1629.?https://doi.org/10.1111/j.1742-4658.2006.05192.x
26.Balajee?AS,?Bohr?VA.?2000.?Genomic heterogeneity of nucleotide excision repair.?Gene.?250(1-2):15-30.?https://doi.org/10.1016/s0378-1119(00)00172-4
27.Gros?L,?Saparbaev?MK,?Laval?J.?2002.?Enzymology of the repair of free radicals-induced DNA damage.?Oncogene.?21(58):8905-8925.?https://doi.org/10.1038/sj.onc.1206005
28.You?J,?Wang?M,?Lee?S.?2003.?Biochemical Analysis of the Damage Recognition Process in Nucleotide Excision Repair.?J. Biol. Chem..?278(9):7476-7485.?https://doi.org/10.1074/jbc.m210603200
29.Rosell?R,?Taron?M,?Barnadas?A,?Scagliotti?G,?Sarries?C,?Roig?B.?2003.?Nucleotide Excision Repair Pathways Involved in Cisplatin Resistance in Non-Small-Cell Lung Cancer.?Cancer Control.?10(4):297-305.?https://doi.org/10.1177/107327480301000404
30.Vogel?U,?Dybdahl?M,?Frentz?G,?Nex??BA.?2000.?DNA repair capacity: inconsistency between effect of over-expression of five NER genes and the correlation to mRNA levels in primary lymphocytes.?Mutation Research/DNA Repair.?461(3):197-210.?https://doi.org/10.1016/s0921-8777(00)00051-3
31.Hanawalt?PC.?2002.?Subpathways of nucleotide excision repair and their regulation.?Oncogene.?21(58):8949-8956.?https://doi.org/10.1038/sj.onc.1206096
32.Critchlow?SE,?Jackson?SP.?1998.?DNA end-joining: from yeast to man.?Trends in Biochemical Sciences.?23(10):394-398.?https://doi.org/10.1016/s0968-0004(98)01284-5
33.Wang?H.?2003.?Biochemical evidence for Ku-independent backup pathways of NHEJ.?Nucleic Acids Research.?31(18):5377-5388.?https://doi.org/10.1093/nar/gkg728
34.Perrault?R,?Wang?H,?Wang?M,?Rosidi?B,?Iliakis?G.?2004.?Backup pathways of NHEJ are suppressed by DNA-PK.?J. Cell. Biochem..?92(4):781-794.?https://doi.org/10.1002/jcb.20104
35.Sánchez-Pérez?I.?2006.?DNA repair inhibitors in cancer treatment.?Clin Transl Oncol.?8(9):642-646.?https://doi.org/10.1007/s12094-006-0034-8
36.Madhusudan?S,?Hickson?ID.?2005.?DNA repair inhibition: a selective tumour targeting strategy.?Trends in Molecular Medicine.?11(11):503-511.?https://doi.org/10.1016/j.molmed.2005.09.004
37.PLUMMER?E.?2006.?Inhibition of poly(ADP-ribose) polymerase in cancer.?Current Opinion in Pharmacology.?6(4):364-368.?https://doi.org/10.1016/j.coph.2006.02.004
38.SABHARWAL?A,?MIDDLETON?M.?2006.?Exploiting the role of O6-methylguanine-DNA-methyltransferase (MGMT) in cancer therapy.?Current Opinion in Pharmacology.?6(4):355-363.?https://doi.org/10.1016/j.coph.2006.03.011
Aladdin:https://www.aladdinsci.com


 危险品化学品经营许可证(带存储)
危险品化学品经营许可证(带存储)